
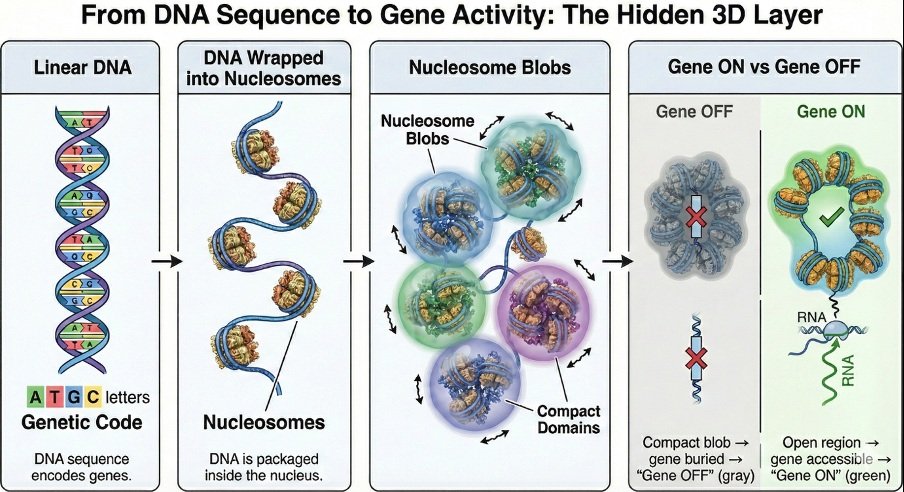
When most of us think of DNA, we imagine a long string of genetic letters A, T, G, and C encoding instructions for life. In reality, DNA inside our cells is not stretched out like a straight thread. Nearly two meters of DNA are tightly packed into a nucleus that is only a few micrometers wide. This raises a question that began to intrigue me deeply: if the DNA sequence is the script of life, what is the stage on which this script is performed? The answer lies not only in the sequence of DNA, but in how it folds. In our recent work, my colleagues and I set out to understand this hidden layer of organization. We demonstrate that DNA is not crumpled chaotically, but rather folded with remarkable precision. Increasingly, we are realizing that this folding is not merely a packaging solution it is part of the regulatory logic of the cell itself.
“The genome is not just a sequence of letters it is a dynamic three-dimensional structure, and the way it folds quietly determines which genes come to life.“
For many years, experimental techniques such as Hi-C and the more refined Micro-C have allowed us to measure which parts of the genome interact with each other inside the nucleus. These powerful methods generate maps of contacts, revealing which genomic regions come into proximity in three-dimensional space. In simple terms, they tell us who “talks” to whom. But they do not directly show us what the actual three-dimensional structure looks like. As a physical chemist working in biology, I felt this was fundamentally a problem of statistical mechanics. DNA is a polymer. Nucleosomes interact. Physical forces govern folding. If we could combine experimental contact maps from Hi-C and Micro-C with physical principles, perhaps we could reconstruct structures consistent with those measurements. There was, however, an important subtlety. Hi-C and Micro-C do not capture a single structure. They measure millions of cells simultaneously, producing contact maps that represent averages over many fluctuating conformations. The data themselves are ensemble-averaged. That realization changed the modeling strategy. Instead of searching for one “correct” structure, we built ensembles collections of many possible conformations that collectively reproduce the experimental contact map. Our models therefore reflect the same statistical nature as the experiments. The genome is not static. It fluctuates. It breathes. It continuously explores different shapes.
And from these reconstructions, a striking pattern emerged. DNA in the nucleus is wrapped around proteins called histones, forming bead-like units known as nucleosomes. When we modeled how these nucleosomes organize in three dimensions, we observed that they tend to cluster into compact groups. We call these clusters “nucleosome blobs.” To visualize this, imagine a long chain of beads loosely folded in your hand. Some beads gather tightly together, forming small clumps, while others remain more extended. These clumps are not permanent they fluctuate yet their statistical presence is robust and reproducible.
What surprised us most was how strongly these blobs correlated with gene activity. Regions that formed tight, stable blobs were often associated with reduced gene expression. In contrast, regions that were more open or fragmented tended to be transcriptionally active. In simple terms, genes do not function only because of what they contain in their sequence. They function depending on the structural neighborhood in which they reside. It is important to note that our modeling does not explicitly simulate every chemical detail such as specific epigenetic marks, protein-binding events, or chromatin-modifying complexes. Yet these effects are not ignored. They are implicitly encoded in the experimental contact maps and nucleosome positioning data that we use as inputs. In other words, the biochemical state of the genome leaves its signature in the interaction patterns we model. Nucleosome positioning, in particular, plays a crucial role. The arrangement of nucleosomes along DNA imposes geometric and entropic constraints. Where nucleosomes are regularly spaced, clustering can emerge more readily; where spacing is irregular the structural landscape changes. Even without modeling each chemical interaction individually, the collective effects of epigenetic regulation are reflected in the physical organization we reconstruct.
This realization shifts our perspective. Gene regulation is not controlled only by biochemical signals or molecular switches. It is also governed by physical organization. When the genome folds, distant elements can become neighbors in three-dimensional space. Regulatory elements can approach or separate from genes. Structural domains can insulate regions or bring them into communication.
From a physics standpoint, these patterns emerge naturally from interactions between nucleosomes under confinement. What appears biologically complex may, at least in part, be governed by surprisingly simple physical principles. For me, this convergence of physics and biology is one of the most beautiful aspects of this research. The same statistical mechanical ideas that describe polymers and complex systems can illuminate how our genome functions. The architecture of the genome is not merely an academic curiosity. When folding patterns are disrupted, the consequences can be profound. Misregulation of chromatin organization has been implicated in cancer, developmental disorders, and aging. If structural domains break down or reorganize incorrectly, genes may be activated at the wrong time or silenced when they should be active. By reconstructing nucleosome-level folding using experimentally informed modeling, we move toward predictive understanding. We can begin to ask: What structural changes accompany disease states? Which regions are structurally stable, and which are susceptible to reorganization? Can we identify architectural vulnerabilities before pathological changes fully manifest? In the future, such insights may contribute to structural biomarkers or therapeutic strategies targeting chromatin organization itself.
Perhaps the most important conceptual shift from our work is this: the genome is not a static object. It is an ensemble a dynamic landscape of possible configurations shaped by physical constraints. Rather than asking, “What is the structure?”, we should ask, “What is the distribution of structures, and what stabilizes them?” This ensemble perspective allows us to connect molecular interactions at the nucleosome scale with gene regulation at the cellular scale. It bridges length scales spanning thousands of times in size from nanometers to micrometers within a unified physical framework. We are only beginning to understand this hidden architecture. Future advances may allow us to map nucleosome-level folding across entire genomes, integrate artificial intelligence with physics-based modeling, and build computational “digital twins” of chromatin in health and disease. In my view, the next frontier in genomics lies not only in reading DNA sequences, but in decoding the geometry that organizes them. Because in the end, life is not governed by sequence alone. It is governed by structure, dynamics, and the laws of physics acting quietly inside every cell. And that hidden architecture may hold answers to some of biology’s deepest questions.






{kind=link}