
This article is currently maintained under temporary RFCSR publication support until 13 June 2026.
Malaria, an ancient disease caused by Plasmodium parasites, remains one of the most significant global public health challenges. Among the various Plasmodial species, Plasmodium falciparum is the major contributor to the lethal complications and mortality associated with malaria. As the existing repertoire of antimalarial drugs has been rendered increasingly obsolete by the relentless evolution of parasite resistance, the discovery of new antimalarials has become critical. A detailed understanding of the malaria parasite survival tactics inside the human host would significantly facilitate the identification of new drug targets thereby accelerating the drug discovery process.
P. falciparum being a digenetic parasite, needs human as a host for its asexual phase of its lifecycle. Infective sporozoite forms of the parasite transmitted from the bite of an infected Anopheles mosquito, initially develops inside the liver hepatic cells and later mature into merozoites to infect the human erythrocytes. This erythrocytic stage is the primary driver of both parasite proliferation and the clinical pathology of malaria. The erythrocytic cycle spans approximately 48 hours, during which the parasite progresses through distinct developmental stages, ultimately yielding multiple daughter merozoites. The subsequent egress and reinvasion of new erythrocytes drive the exponential increase in parasite biomass, directly correlating with malaria’s clinical severity.
As with any intracellular parasites, P. falciparum needs to extensively remodel the host human erythrocyte in order to create a suitable niche for their survival. Following erythrocyte invasion, the parasite surrounds itself within a parasitophorous vacuolar membrane (PVM) and constructs a specialized tubulovesicular network. This parasite-derived membrane system serves as a vital conduit, trafficking parasite encoded proteins into the host cell to facilitate survival and pathogenesis. It is believed that around 20% of the P. falciparum erythrocytic proteome cross the PVM to remodel the human erythrocyte. The changes optimize nutrient exchange through increased permeability, modify erythrocyte rigidity through cytoskeletal remodelling, and establish surface knobs that facilitate cytoadherence to prevent splenic clearance of infected erythrocytes.
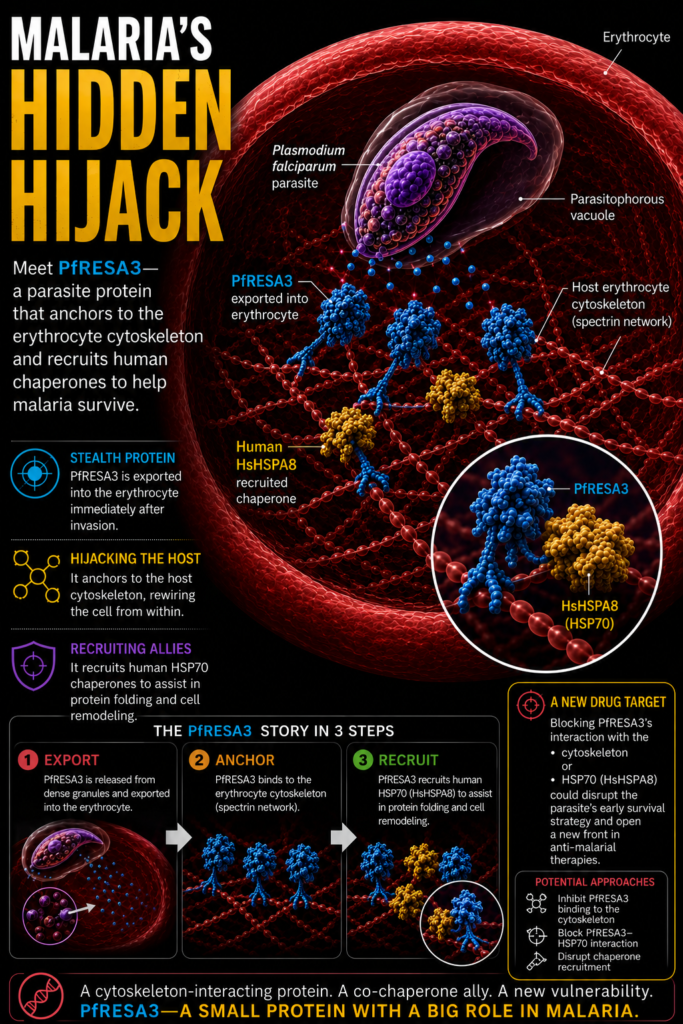
An important aspect of this protein trafficking is that in order to cross the parasite PVM, the proteins have to go through a protein translocon complex (PTEX) where they unfold and get released into the erythrocyte cytosol in unfolded state. For these exported proteins to function and reach their targets in the red blood cell, they must fold correctly; a process heavily dependent on chaperone assistance. Export motif analysis has identified a group of P. falciparum J domain proteins (JDPs) are trafficked into the host erythrocyte, where they likely facilitate protein refolding and trafficking. JDPs characterised by the presence of a conserved J domain with an HPD motif, are not chaperones themselves. Instead, they act as co-chaperones that deliver substrates to HSP70 proteins and activate their folding activity. Curiously, P. falciparum exports only one HSP70 protein (PfHSP70-x), which is dispensable for the parasite survival. In contrast, several of these exported JDPs have been reported to be indispensable for the parasite growth, and thus led to a speculation that human chaperones may be involved in the chaperoning process.In this regard our lab has been working on characterising these JDPs. Our recent report talks about the P. falciparum Ring-infected Erythrocyte Surface Antigen 3 (PfRESA3), identifying it as a dense granule merozoite protein that is exported to the inner face of the host erythrocyte membrane immediately after erythrocyte invasion. Unlike many canonical JDPs, though PfRESA3 lacks the conserved HPD motif, it remains functionally active, acting as a potential co-chaperone. We demonstrated that PfRESA3 interacts with the host cytoskeleton and effectively stimulates the ATPase activity of human HSPA8 (Hsc70). PfRESA3 interaction with the erythrocyte cytoskeleton suggests it to play an important role in erythrocyte cytoskeleton remodelling. By recruiting host chaperones to assist in protein folding, PfRESA3 plays a vital role in the initial ‘hijacking’ of the erythrocyte, providing a molecular blueprint for how the parasite exploits host machinery to ensure its survival.
Our group previously established that P. falciparum JDPs, such as PfA8iJp and PfeCiJp, hijack host erythrocyte chaperones; however, these proteins are primarily trafficked during the later trophozoite and schizont stages (beyond 24 hours post-invasion). Since the extensive remodelling of the host cell begins immediately after invasion, a critical knowledge gap remained regarding how early exported proteins are folded and functionalized. Our recent characterization of PfRESA3 addresses this lacuna. As a dense granule protein exported immediately post-invasion, PfRESA3 hijacks the host chaperone HSPA8, providing the necessary folding machinery to facilitate the parasite’s initial establishment and survival within the erythrocyte.The identification of PfRESA3 as a functional co-chaperone offers a strategic advantage for antimalarial drug discovery by targeting the parasite’s proteostasis network. Because PfRESA3 is exported immediately post invasion, inhibiting its function could stall the remodelling process. Since PfRESA3 stimulates human HSPA8 (Hsc70) without the classic HPD motif, the interaction interface is likely unique compared to human J-domain proteins. Designing small molecules or peptide mimics to block this molecular interaction would prevent the parasite from hijacking host machinery, leaving its exported proteome unfolded and non-functional. Furthermore, molecules that will prevent PfRESA3 from anchoring to the host cytoskeleton may collapse or weaken the scaffold needed for establishing nutrient channels and cyto-adherent knobs. By preventing the formation of these knobs, the infected erythrocyte remains flexible, ensuring it is recognized and cleared by the spleen. This approach of targeting a parasite-specific recruiter of host chaperones would minimize the potential toxicity to human cells while effectively neutralizing the parasite’s ability to survive and sequester within the microvasculature. Furthermore, if this hijacking mechanism is conserved across other human infecting species like P. vivax, targeting PfRESA3 like proteins could pave the way for broad spectrum antimalarial therapeutics.


")






{kind=link}