
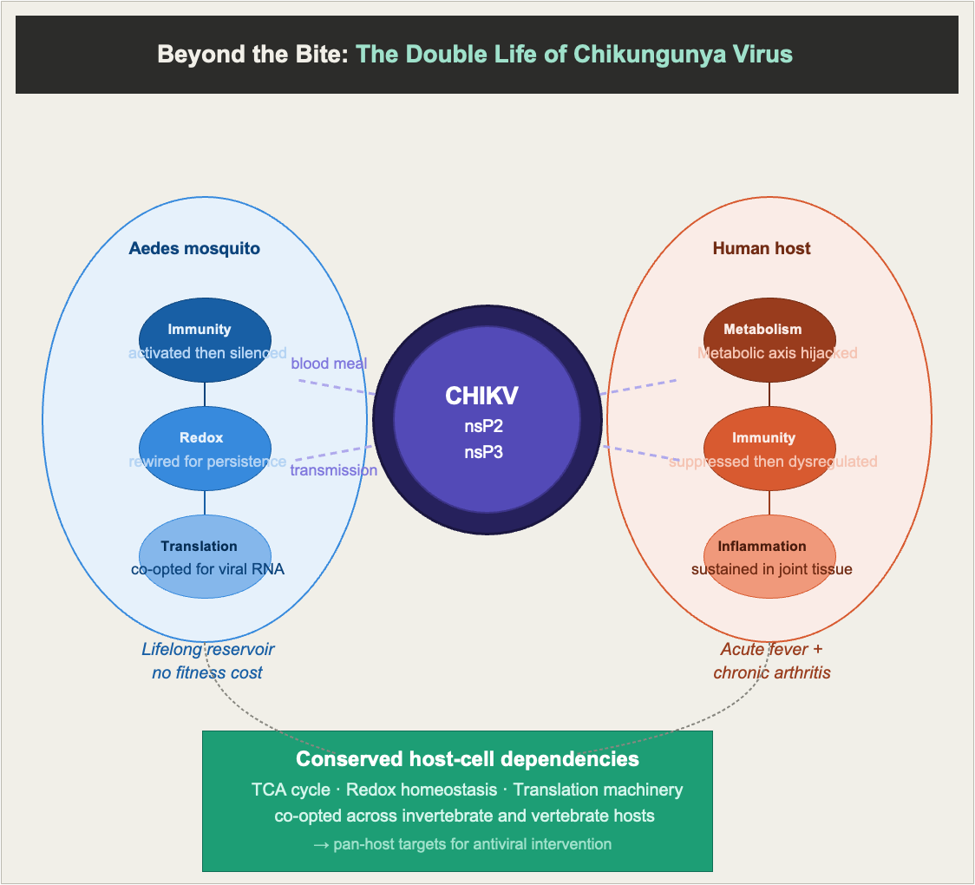
Every time an Aedes mosquito takes a blood meal, it does more than leave an itchy welt. If the mosquito carries an arbovirus such as chikungunya virus (CHIKV) or Dengue virus (DENV), it deposits the pathogen into human skin, triggering a cascade of immune responses that the virus must evade to establish itself. What is remarkable is how differently the virus behaves in the two hosts it depends on: in the mosquito, it persists silently for life without causing harm, while in the human body, it replicates aggressively across multiple tissues. My laboratory at the Vector Borne Diseases Group, ICGEB New Delhi, has spent several years examining both sides of this double life succeeding in two biologically distinct organisms through a conserved set of host–virus interactions.
“In Aedes mosquitoes, CHIKV exploits the vector’s own antioxidant response to silence innate immunity — explaining why mosquitoes remain lifelong viral reservoirs despite mounting a genuine immune defence.”
The tolerant host: Persistent infection without pathology
When CHIKV enters Aedes aegypti, the mosquito mounts an innate immune response centred on reactive oxygen species (ROS). Transcriptomic profiling of infected Aag2 cells showed that early ROS spikes activate the Imd immune pathway through its transcription factor Rel2 — silencing Rel2 increased viral titres approximately 14-fold. However, the escalating ROS also induces antioxidant gene expression — glutathione metabolism, ascorbate synthesis, and cytochrome P450-dependent redox homeostasis — that progressively quenches the very ROS that drive immunity. As oxidative stress decreases, Rel2 signalling declines, relieving the virus of immune pressure and allowing replication to peak around 24 hours post-infection. The mosquito’s own antioxidant self-preservation inadvertently provides the window the virus needs.
A complementary study in whole Aedes aegypti mosquitoes showed that dietary L-cysteine supplementation restricted CHIKV infection, coinciding with elevated glutathione S-transferase activity and reduced oxidative damage. Silencing genes in the taurine and hypotaurine biosynthetic pathway further altered viral loads and redox biology — confirming the cysteine–antioxidant metabolic axis as a genuine determinant of how much virus a mosquito harbours and transmits.
Viral non-structural proteins: Master host manipulators
CHIKV encodes four non-structural proteins (nsP1–nsP4) that form the replication complex, but each also recruits and repurposes host proteins. Our group systematically mapped the interactomes of nsP2 and nsP3 across human liver cells (Huh7), macrophages (THP-1), and Aedes albopictus cells, producing the most comprehensive cross-host interaction dataset for any CHIKV non-structural protein. In human cells, these viral proteins engage host networks enriched for TCA cycle enzymes, translation regulators, proteostasis machinery, and immune signalling through NF-κB and cGMP-PKG pathways. For nsP2, a direct comparison across human Huh7 and Aedes U4.4 cells identified six conserved interactors shared between both hosts — DEAD-box helicase, eIF3, succinate dehydrogenase, isocitrate dehydrogenase, glutathione S-transferase, and peptidyl-prolyl isomerase. This conserved core defines the pan-host dependencies CHIKV exploits in organisms as different as a mammal and a mosquito.
The infected cell: metabolic reprogramming across tissues
In human liver cells, argininosuccinate synthase 1 (ASS1), an enzyme of the urea cycle, emerged as an unexpected proviral factor. ASS1 sits at a metabolic fork where arginine can either feed nitric oxide synthase (NOS) to generate antiviral nitric oxide, or be converted by arginase 1 (ARG1) to ornithine and polyamines that support viral replication. CHIKV shifts this balance in its favour: ASS1 and ARG1 are upregulated during infection, polyamine levels rise, and nitric oxide falls. Silencing ASS1 reversed these changes and reduced viral titres by over 95%. ASS1 also suppresses STAT3, a key antiviral signalling protein, during infection — a suppression restored upon ASS1 silencing. Notably, succinate dehydrogenase and isocitrate dehydrogenase also appear as conserved nsP2 interactors across human and mosquito cells, suggesting convergent targeting of the TCA cycle is a signature of how CHIKV manages energy metabolism in every host.
In the joint, chikungunya disease causes arthritis that can outlast the acute infection by months or years. Standard rodent models fail to recapitulate this chronicity. Our group developed a three-dimensional spheroid system from primary human chondrocytes that sustains productive CHIKV infection for up to seven days. Global proteomics of infected spheroids revealed activation of interferon-stimulated genes, NF-κB inflammatory signalling, elevated IL-6, TNF-α, and IL-1β, and perturbation of arginine and proline metabolic pathways — echoing the liver findings. The sustained inflammatory response, with matrix metalloprotease activity maintained over days, appears to precisely drive extracellular matrix remodelling and chronic joint damage. The enemy is partly the virus, but partly the cell’s own prolonged attempt to fight it.
Across the two hosts: Conserved host dependencies
Across every system studied — mosquito cells, human liver, joint, and macrophages — CHIKV converges on the same molecular themes: TCA cycle enzymes, redox homeostasis, and translation initiation machinery. This convergence is encouraging therapeutically, because it suggests a small set of host-directed interventions could interrupt CHIKV replication across tissues and potentially in the vector. Unlike direct antivirals, host-directed strategies targeting metabolic enzymes or redox pathways are inherently harder for a fast-evolving RNA virus to escape through mutation.
Challenges and the road ahead
Significant challenges remain. Pharmacological inhibitors suitable for clinical use do not yet exist for key host targets such as ASS1. The in vitro systems used, however sophisticated, do not fully capture immune cell crosstalk and systemic metabolism in a living organism. Validation in in vivo models and functional perturbation of conserved interactors are important next steps. Extending the interactome approach to nsP1 and nsP4 would complete the replication complex map. On the vector side, translating the redox and taurine–cysteine findings into strategies that reduce mosquito vector competence is a long-term goal with direct public health impact.
“Comparative interactome mapping of CHIKV nsP2 in human and mosquito cells uncovered six conserved host proteins spanning TCA cycle enzymes, translation factors, and redox regulators — defining pan-host dependencies the virus exploits across species.”
Looking forward
Our findings suggest CHIKV infection is better understood as a sequence of tissue-specific negotiations, each conducted using a similar vocabulary of metabolic and immune signalling molecules shaped by the virus’s two-host evolutionary history. The mosquito is not merely a syringe; it is a co-evolutionary partner that has shaped which host vulnerabilities CHIKV exploits. Understanding both sides of this relationship simultaneously may be the most efficient path to interventions that work at the source — in the vector — as well as at the site of disease in the human patient.
A concluding message
Chikungunya virus is a master of metabolic persuasion. In the mosquito, it outwits immunity by letting the vector’s antioxidant defences suppress the very signals that would clear it. In humans, it hijacks arginine metabolism, redirects TCA cycle energy, co-opts translation machinery, and sustains inflammatory signalling in joints long after active replication has waned. The same non-structural proteins accomplish all of this across both hosts — a finding that points toward shared therapeutic targets and a more unified vision of arboviral biology.










{kind=link}